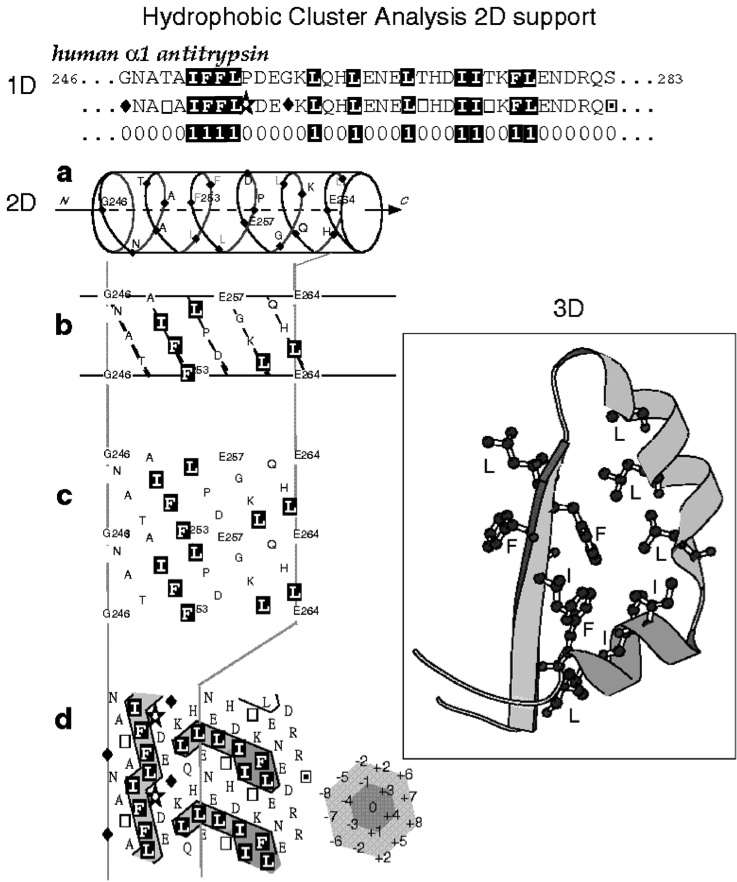
Hydrophobic Cluster Analysis (HCA) is based on a two-dimensional representation of the protein sequence, in which hydrophobic amino acids congregate into clusters (Callebaut, et al., 1997; Gaboriaud, et al., 1987; Figure 1). Statistical studies performed on experimental 3D structures have shown that hydrophobic clusters mainly correspond to regular secondary structures, and have supported the relevance of the chosen hydrophobic alphabet, as well as the alpha-helix as 2D support for revealing this structural information (Woodcock, et al., 1992).
The two-dimensional support dictates the segmentation rules of a sequence into
clusters, exactly as spacers separate words in a text. Hence, two hydrophobic
amino acids participate in two distinct clusters, if they are separated by at
least 4 four non-hydrophobic amino acids or a proline, in the case of an alpha-helical
support. This minimal number of non-hydrophobic amino acids is called the connectivity
distance and is linked to the distance separating an amino acid from its furthest
close neighbor. The 2D support is thus a convenient mean for revealing the 2D
neighborhood of each amino acid. Hydrophobic clusters, which are binary patterns
constrained by the connectivity distance, are much more informative than simple
binary patterns as they allow to reveal the 2D context in which the binary pattern
is embedded (Hennetin, et al., 2003). Approximately 200 different species of
hydrophobic clusters (a species corresponding to a given constrained binary
pattern), which bring together a large part of the total number of hydrophobic
clusters, have strong tendencies towards alpha helices or beta strands (Eudes,
et al., 2007).
HCA is often considered as an approach allowing the prediction of secondary
structures from the only knowledge of a protein sequence. It however allows
to combine this prediction with the comparison of 1D sequences, which makes
it a powerful tool for helping the identification of remote relationships.
Identification of repeated sequences is an immediate output of the analysis
of HCA plots (see 1 for references, Figure 2), but HCA can also be used for
identifying relevant 2D signals in the non significant results provided by current
similarity search methods. In both cases, amino acid identities and similarities
can be put at the level of the HCA plots and evaluated in the context of the
secondary structure content. The shapes of hydrophobic clusters, containing
the hydrophobic amino acids for which hydrophobicity is conserved in the 1D
alignment, can be compared and put into perspective to the minimal hydrophobic
core positions, which can be defined from a set of homologous sequences (Poupon
and Mornon, 1998). The ability of HCA to identify remote relationships relies
on the evolutive robustness of hydrophobic clusters, as compared to 1D sequences,
but also on the independence of the method relative to indels, which can be
large and limit the alignment of related sequences to only a small part of the
domain (Figure 3).
The wide-range application of the method has so far been limited by the need of human expertise. Tools are however being developed to facilitate its use (dictionary of hydrophobic clusters (Eudes et al., 2007), automatic HCA-based delineation of globular like-domain (Faure and Callebaut, submitted for publication), HCA plot display within sequence similarities searches, together with domain architecture information (Faure and Callebaut, 2013), ....
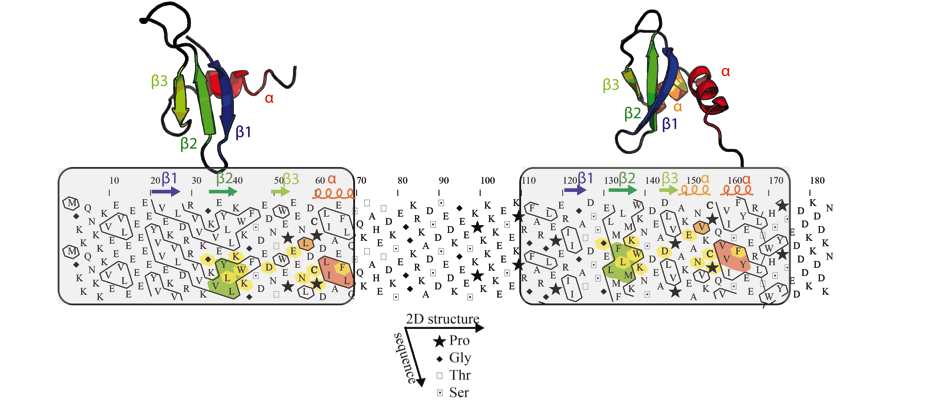
Figure 2: Example of the detection of an internal duplication
in the mouse chromobox homolog 1 (CBX1_MOUSE, UniProt P83917).
Globular domains (boxed), containing approximately one third of hydrophobic
amino acids gathered into clusters, are separated by a hinge, which is clearly
less hydrophobic. The comparison of the HCA plots of the two domains indicates
similar shapes of clusters (shaded green and red), suggesting a structural relationship.
This potential relationship is further strengthened by sequence identities (shaded
yellow) identified relative to the conserved positions of hydrophobic amino
acids within clusters and is supported here at the 3D level (the observed 2D
and 3D structures of the chromo and chromo-shadow domains of this protein are
shown up to the HCA plot (pdb identifiers: 1AP0 and 1DZ1, respectively)). The
hydrophobic clusters shaded in green and red correspond to the internal strand
beta 2 and to the C-terminal alpha helix, respectively. In the chromo shadow
domain, the loop linking strand beta3 and the C-terminal alpha helix includes
a short alpha helix, containing two alanine residues.
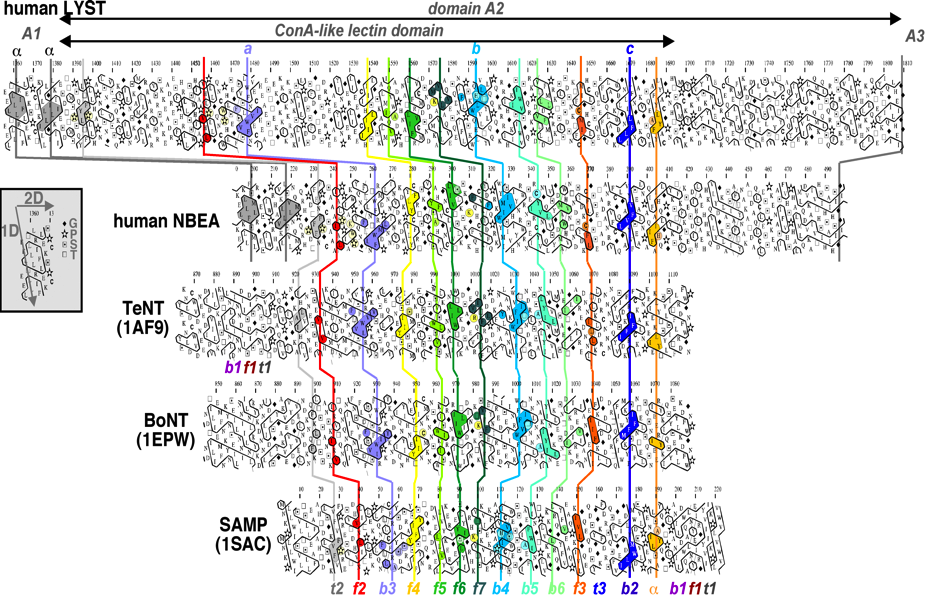
Figure 3: Example of a HCA alignment with large indels. Comparison of the HCA plots of BEACH domains A2 with ConA-like lectin domains, whose 3D structures are known (extracted from (Burgess, et al., 2009)).
Footnotes
1. The first published example relies on the identification of a repeated domain in the extracellular region of cytokine receptors (Thoreau et al., 1991) and several other examples have illustrated since this ability for detecting repeated domains (e.g. BRCT domains in BRCA1 (Callebaut & Mornon, 1997a), BAH domains in DNA methyltransferases (Callebaut et al., 1999), Chromo and Chromo-shadow domains in HP1 (Ye et al., 1997), LEM domains in inner nuclear membrane proteins (Laguri et al., 2001, Lin et al., 2000) and ZP-N domains in Zona Pellucida proteins (Callebaut et al. 2007) or repeated motifs (e.g. blades of beta propellers in RAG2 (Callebaut & Mornon, 1998) and in Sec12 (Callebaut & Chardin, 2002), OCRE imperfect repeats (Callebaut & Mornon, 2005)).
2. e.g. FKBP domain in trigger factor (Callebaut & Mornon, 1995), GTFs in P.falciparum (Callebaut et al. (2005), relationship of Cernunnos to Nej1/XRCC4 (Callebaut et al., 2006), a concanavalin A-like lectin domain in CHS1/LYST (Burgess et al. , 2009).
3. e.g. BRCT (Callebaut & Mornon, 1997a), TUDOR (Callebaut & Mornon, 1997b), BAH (Callebaut et al., 1999), RUN (Callebaut et al., 2001), LOTUS (Callebaut & Mornon, 2010).
4. e.g. Glycoside hydrolases (Henrissat et al., 1995), hydrolases of the beta-CASP family of metallo-enzymes (Callebaut et al., 2002).
References
• Burgess, A., et al. (2009) A concanavalin A-like lectin domain in the
CHS1/LYST protein, shared by members of the BEACH family., Bioinformatics, 25,
1219-1222.
• Callebaut, I. and Mornon, J.P. (1995) Trigger factor, one of the Escherichia
coli chaperone proteins, is an original member of the FKBP family., FEBS Lett.
, 374, 211-215.
• Callebaut, I., et al. (1997) Deciphering protein sequence information
through hydrophobic cluster analysis (HCA): current status and perspectives,
Cell. Mol. Life Sci., 53, 621-645.
• Callebaut, I. and Mornon, J.P. (1997a) From BRCA1 to RAP1: a widespread
BRCT module closely associated with DNA repair., FEBS Lett, 400, 25-30.
• Callebaut, I. and Mornon, J.P. (1997b) The human EBNA-2 coactivator
p100: multidomain organization and relationship to the staphylococcal nuclease
fold and to the tudor protein involved in Drosophila melanogaster development,
Biochem J, 321, 125-132.
• Callebaut, I. and Mornon, J.P. (1998) The V(D)J recombination activating
protein RAG2 consists of a six-bladed propeller and a PHD fingerlike domain,
as revealed by sequence analysis., Cell Mol Life Sci, 54, 880-891.
• Callebaut, I., Courvalin, J.C. and Mornon, J.P. (1999) The BAH (bromo-adjacent
homology) domain: a link between DNA methylation, replication and transcriptional
regulation., FEBS Lett, 446, 189-193.
• Callebaut, I., et al. (2001) RUN domains: a new family of domains involved
in Ras-like GTPase signaling., Trends Biochem Sci, 26, 79-83.
• Callebaut, I., et al. (2002) Metallo-beta-lactamase fold within nucleic
acids processing enzymes: the beta-CASP family., Nucleic Acids Res, 30, 3592-3601.
• Callebaut, I. and Chardin, P. (2002) The yeast Sar exchange factor Sec12,
and its higher organism orthologs, fold as beta-propellers., FEBS Lett, 525,
171-173.
• Callebaut, I., et al. (2005) Prediction of the general transcription
factors associated with RNA polymerase II in Plasmodium falciparum: conserved
features and differences relative to other eukaryotes., BMC Genomics, 6, 100.
• Callebaut, I. and Mornon, J.P. (2005) OCRE: a novel domain made of imperfect,
aromatic-rich octamer repeats., Bioinformatics, 21, 699-702.
• Callebaut, I., et al. (2006) Cernunnos interacts with the XRCC4 x DNA-ligase
IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1.,
J Biol Chem, 281, 13857-13860.
• Callebaut, I., Mornon, J.P. and Monget, P. (2007) Isolated ZP-N domains
constitute the N-terminal extensions of Zona Pellucida proteins., Bioinformatics,
23, 1871-1874.
• Callebaut, I. and Mornon, J.P. (2010) LOTUS, a new domain associated
with small RNA pathways in the germline., Bioinformatics, 26, 1140-1144.
• Eudes, R., et al. (2007) A generalized analysis of hydrophobic and loop
clusters within globular protein sequences, BMC structural biology, 7, 2.
• Faure, G. and Callebaut, I. (2013) Identification of hidden relationships
from the coupling of Hydrophobic Cluster Analysis and Domain Architecture information,
Bioinformatics, in press.
• Gaboriaud, C., et al. (1987) Hydrophobic cluster analysis: an efficient
new way to compare and analyse amino acid sequences., FEBS Lett, 224, 149-155.
• Hennetin, J., et al. (2003) Non-intertwined binary patterns of hydrophobic/nonhydrophobic
amino acids are considerably better markers of regular secondary structures
than nonconstrained patterns., Proteins, 51, 236-244.
• Henrissat, B., et al. (1995) Conserved catalytic machinery and the prediction
of a common fold for several families of glycosyl hydrolases., Proc Natl Acad
Sci U S A, 92, 7090-7094.
• Laguri, C., et al. (2001) Structural characterization of the LEM motif
common to three human inner nuclear membrane proteins., Structure, 9, 503-511.
• Lin, F., et al. (2000) MAN1, an inner nuclear membrane protein that
shares the LEM domain with lamina-associated polypeptide 2 and emerin., J Biol
Chem, 275, 4840-4847.
• Poupon, A. and Mornon, J.P. (1998) Populations of hydrophobic amino
acids within protein globular domains: identification of conserved "topohydrophobic"
positions., Proteins, 33, 329-342.
• Thoreau, E., et al. (1991) Structural symmetry of the extracellular
domain of the cytokine/growth hormone/prolactin receptor family and interferon
receptors revealed by hydrophobic cluster analysis., FEBS Lett. , 282, 26-31.
• Woodcock, S., Mornon, J.P. and Henrissat, B. (1992) Detection of secondary
structure elements in proteins by hydrophobic cluster analysis., Protein Eng,
5, 629-635.
• Ye, Q., et al. (1997) Domain-specific interactions of human HP1-type
chromodomain proteins and inner nuclear membrane protein LBR., J Biol Chem.,
272, 14983-14989.