Jacques Chomilier
Senior Scientist at
CNRS


Backgound
Solid State Physics: electron
densities; synchrotron; X-Ray inelastic scattering.
Structural
Bioinformatics: Monte-Carlo simulation; protein folding nucleus; stability towards
mutation; sub-domain fragments; protein peptide interactions.
Research
Protein
folding simulation
Globular
proteins can be described from a physical point of view as micelles with a
hydrophobic core surrounded by a hydrophilic shell. The process driving the
formation of a compact globule after the synthesis of the chain of amino acids is
not fully understood. Simulations can be proposed, provided major
approximations are done. One of them realises in transformation of the
continuous space into a discrete space, where the amino acids are placed at the
nodes of a lattice. The starting conformation is then chosen from a random
trial. Successive displacements are performed on the amino acids, by means of
the Monte-Carlo selection of the final state.
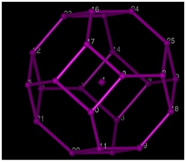
Lattice
used to simulate the folding of proteins. Spheres represent the positions
allowed to the amino acids.
During
the simulation, the number of non-covalent neighbours is periodically recorded
and the mean is calculated at the end of the process. It results peaks in this
distribution, and the maxima are called the MIR (Most Interacting Residues). It
has been shown over the past years that these MIR are key residues, not for the
function, but for the structure, and deeply buried in the core of the protein.
A web server is available for MIR prediction, at RPBS: http://sprouts.rpbs.univ-paris-diderot.fr/mir.html
Fragments
of globular proteins
A
globular protein is classically presented, from a structural point of view, as
a succession of regular secondary structures (strands and helices) connected by
loops. One can ask the question of the building blocks used by nature to
provide a given structure to a sequence. These elementary bricks have to be
stable on their own, and one of the assumptions is to consider the super
secondary structure level, corresponding to a typical number of amino acids of
the order of 20-25. By searching increased stability by the fact that the ends
of the constituting fragments are close one from each other in the 3D space,
results in the concept of TEF (Tightened End Fragments). Also the splitting of
one protein into TEF is not unique, one can propose an algorithm that biases
the search towards fragments with the extremities into the core of the protein.
It has been shown that these ends are mainly occupied by hydrophobic residues
(any of the list Ile, Leu, Met, Phe,
Trp, Tyr, Val). They can be used to drive the search
for inhibitors of protein protein interactions. TEF
calculation is available at http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py
Stability
with respect to point mutation
Stability
of a position by respect to mutation can be done by comparing the free energies
of the native and mutated proteins, DDG. Several tools are available,
either as included in a web server, or as an executable file. They are not
completely coherent as results, and they have been placed together on a web
server, called SPROUTS, accessible
at http://sprouts.rpbs.univ-paris-diderot.fr.
If one transforms the DDG values from kcal/mol into
scores, ranging from 0 (no effect) to 19 (completely destabilizing or
stabilizing), it appears that the extrema are fairly well located at the same position,
regardless of the algorithm used. For the moment, the algorithm available are: Imutant (various versions), Dfire,
PopMuSiC, Fold-X, Mupro. A
mutation is considered as significant if the free energy differs from the
native one by more than 2 kcal/mol.
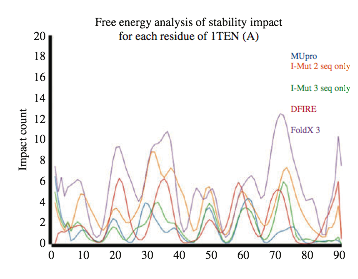
Example of
the stability impact of mutations for the 19 possible side chain substitutions,
at each position, for five algorithms.
Publications (selection)
Contribution to the prediction of
the fold code: application to immunoglobulin
and flavodoxin. M. Banach, N. Prudhomme, M.
Carpentier, E. Duprat, N. Papandreou, B. Kalinowska,
J. Chomilier, I. Roterman. Plos One (2015) 10:e0125098
Prediction of stability upon point mutation in the context of the folding nucleus. M. Lonquety, J. Chomilier, N. Papandreou, Z. Lacroix. OMICS (2010) 14: 151-156
Prediction of the protein folding core: application to the immunoglobulin
fold. N. Prudhomme, J. Chomilier.
Biochimie (2009) 91:1465-1474
SPROUTS: a database for evaluation of the protein stability upon point mutation. M. Lonquety, J. Chomilier, N. Papandreou, Z. Lacroix. Nucleic Acids Res (2009) 37:D374-D379
Tandem duplication of a degeneLast
printed 00/00/00
00:00rated GTP-binding domain at the origin
of GTPase receptors Toc159 and thylakoidal
SRP. J. Hernandez Torres, M. Arias Maldonado, J. Chomilier.
BBRC (2007) 364:325-331
RPBS: a web resource for structural bioinformatics. C. Alland, F. Moreews, D. Boens, M. Carpentier, S. Chiusa, M. Lonquety, N. Renault, Y. Wong, H. Cantalloube, J. Chomilier, J. Hochez, J. Pothier, B. Villoutreix, J.-F. Zagury, P. Tufféry. Nucleic Acids Res. (2005) 33:W44-W49
Improvement of collagen-induced arthritis by active immunization against murine Il1 peptides designed by molecular modelling. S. Bertin-Maghit, C. Capini, N. Bessis, J. Chomilier, S. Muller, A. Abbas, L. Autin, J.-L. Spadoni, J. Rappaport, A. Therwath, M.C. Boissier, J.F. Zagury. Vaccine (2005) 23:4228-4235
Universal positions in globular proteins : observation to simulation. N. Papandreou, E. Eliopoulos, I Berezovsky, A. Lopes, E. Eliopoulos, J. Chomilier. Eur. J. Biochem. (2004) 271:4762-4768
Identification of two immunogenic domains of the prion protein –PrP- which activate class II restricted T cells and elicit antibody responses against the native molecule. S. Grégoire, C. Logre, P. Metharom, E. Loing, J. Chomilier, M. Bruley Rosset P. Aucouturier, C. Carnaud. (2004) J. Leukoc. Biol. 76 125-134
Active immunization
against murine TNF peptides in mice:
generation of autologous antibodies cross-reacting with the native cytokines. C. Capini,
S. Bertin-Maghit, P. Haumont,
E. Bernier, E. Muel, M. Laborie, L. Autin, S. Paturance, N. Bessis, M.C. Boissier, J. Chomilier,
P. Cohen, A. Therwath, J.F. Zagury.
(2004) Vaccine 22:3144-3453
Nonatomic solvent driven Voronoï tessellation of proteins: an open tool to analyze protein folds. B. Angelov, J.-F. Sadoc, R. Jullien, A. Soyer, J.-P. Mornon, J. Chomilier. Proteins (2002) 49:446-452
Distribution of tightened end fragments of globular proteins statistically match that of topohydrophobic positions: towards an efficient punctuation of protein folding? M. Lamarine, J.-P. Mornon, I. N. Berezovsky, J. Chomilier. Cell. Mol. Life sci. (2001) 58:492-498
Voronoi tessellation reveals the condensed matter character of folded proteins. A. Soyer, J. Chomilier, J.-P. Mornon, R. Jullien, J.-F. Sadoc. Phys. Rev. Lett. (2000) 85 3532-3535
Beta sheet modeling by helical surfaces. D. Znamenskiy, K. Le Tuan, A. Poupon, J. Chomilier, J.-P. Mornon. Prot. Enging. (2000) 13 407-412
New efficient statistical sequence dependent structure prediction of short to medium-sized protein loops based on an exhaustive loop classification. J. Wojcik, J.-P. Mornon, J. Chomilier. J. Mol. Biol. (1999) 289 1469-1490
Molecular modeling of immunoglobulin light chains implicates hydrophobic residues in non-amyloid light chain deposition disease. S. Déret, J. Chomilier, D.-B. Huang, J.-L. Preud'homme, F. J. Stevens, P. Aucouturier. Prot. Enging. (1997) 10 1191-1197
Deciphering protein sequence information through Hydrophobic Cluster Analysis (HCA) : current status and perspecrives. I. Callebaut, G. Labesse, P. Durand, A. Poupon, L. Canard, J. Chomilier, B. Henrissat, J.-P. Mornon. Cell. Mol. Life sci. (1997) 53 621-645. Article de revue
A global taxonomy of loops in globular proteins. J.-M. Kwasigroch, J. Chomilier, J.-P. Mornon. J. Mol. Biol. (1996) 259 855-872
Teaching (present)
Molecular
Modelling, M2 In Silico Drug Desgin,
Paris Diderot University
Collaborations (present)
Zoé Lacroix, Arizona
State University, Tempe, USA
Nikolaos Papandreou, Agricultural University
Athens, Greece
Irena Roterman, Jagiellonian University, Krakow, Poland
Contact
IMPMC, Université Pierre & Marie Curie
Tour 23-22
Room 408
4 Place Jussieu
75 Paris
France
Tel: 33 +1
44 27 50 79
Fax: 33 +1
44 27 37 85
Mail:
chomilier@impmc.jussieu.fr